
d-SEAMS
Deferred Structural Elucidation Analysis for Molecular Simulations
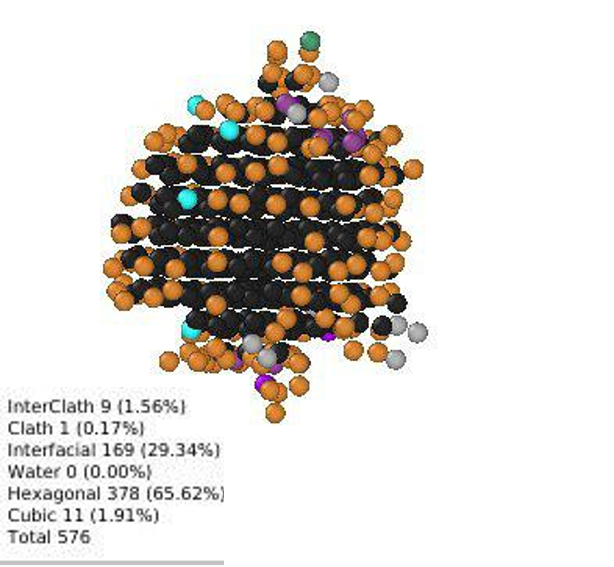
Representative image of various ice types (Ic, Ih and Interfacial) forming on a surface
About
Developed actively at the Computational Nano Science Group of IIT Kanpur, under the aegis of Jayant K. Singh and is being prepared for publication.
d-SEAMS is a collection of analysis tools for molecular simulations. It has been implemented as a High Performance Cluster enabled engine in C++, with extensions via the Lua scripting interface. The code sheds light on the structures from simulation trajectories, in terms of their topology and it has been tailored for the analysis of nucleating systems. The framework is meant to be interfaced to a large variety of external tools and software, including R and other libraries. It is also the first and at-present only scientific analysis tool to use reproducible builds via nix.
The full documentation and installation instructions are here: https://docs.dseams.info
Citation
This has been published at the Journal of Chemical Information and Modeling (JCIM)
You may also read the preprint on arXiv
If you use this software please cite the following:
Goswami, R., Goswami, A., & Singh, J. K. (2020). d-SEAMS: Deferred Structural Elucidation Analysis for Molecular Simulations. Journal of Chemical Information and Modeling. https://doi.org/10.1021/acs.jcim.0c00031Software Features
d-SEAMS, as a toolkit has features intended both for long-term mantainance and extensibility, but also high performance insightful execution.
Plugin Overview
d-SEAMS, as a scientific tool was initially designed to overcome analysis bottlenecks for nucleating systems simulated with LAMMPS.
First Installation
The documentation and README are very detailed, but given the nature of the nix setup, the following first installation video is pertinent and serves as a brief tutorial.
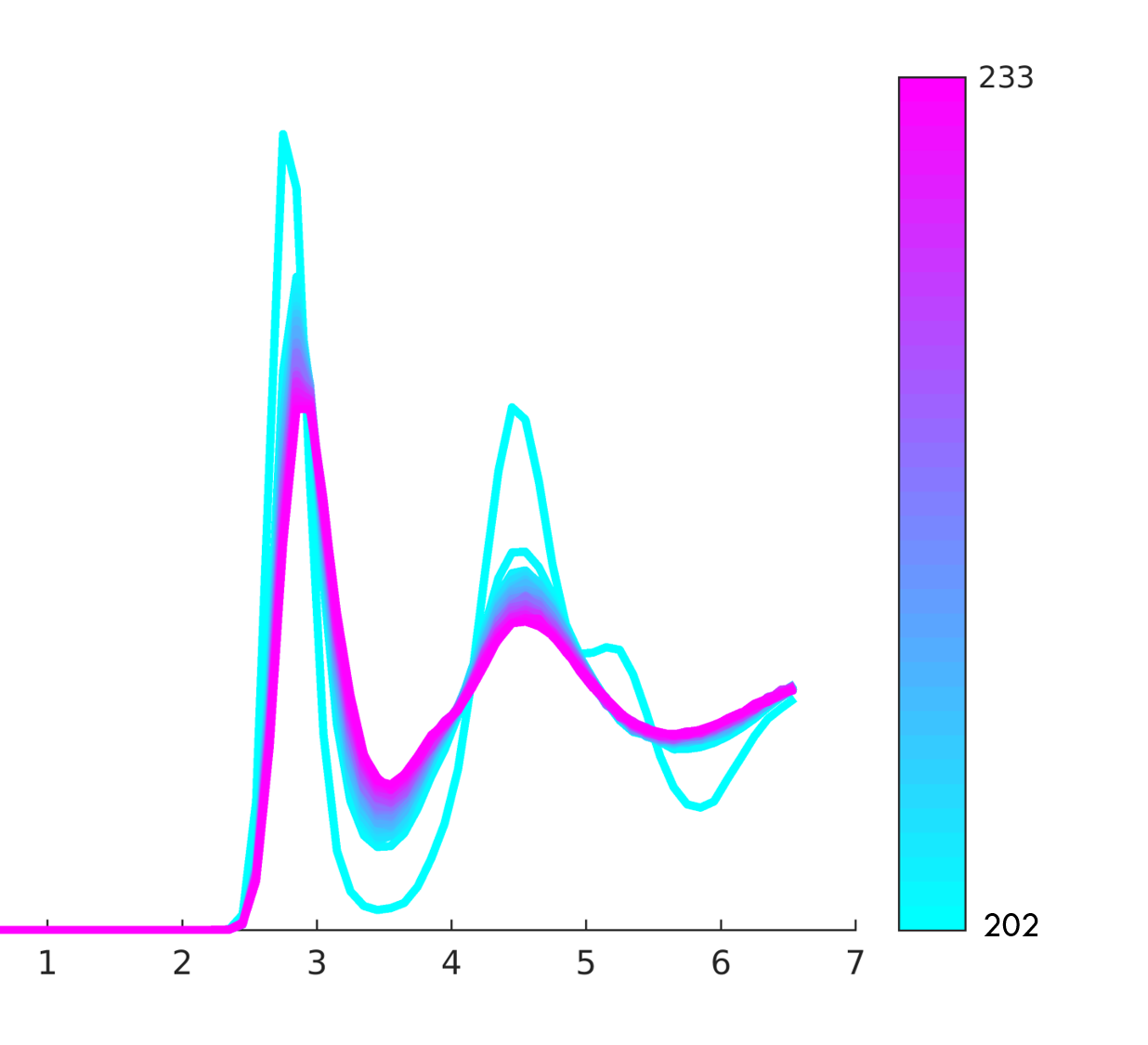
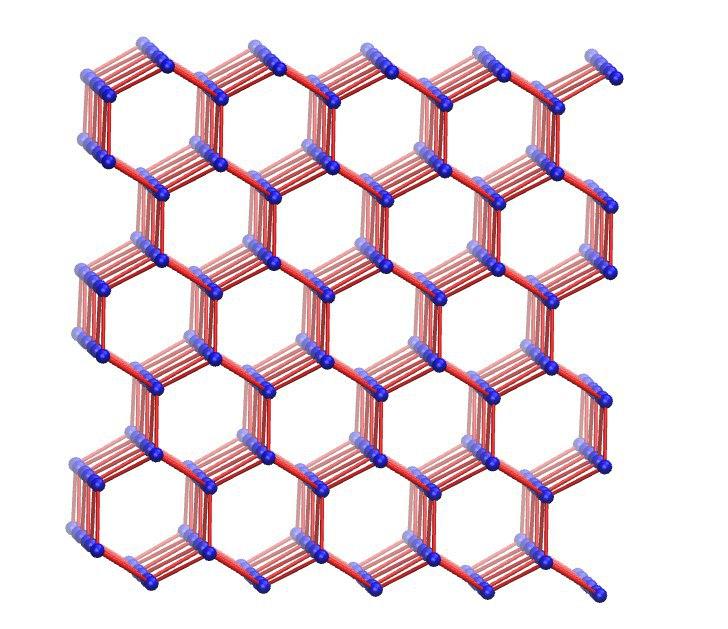
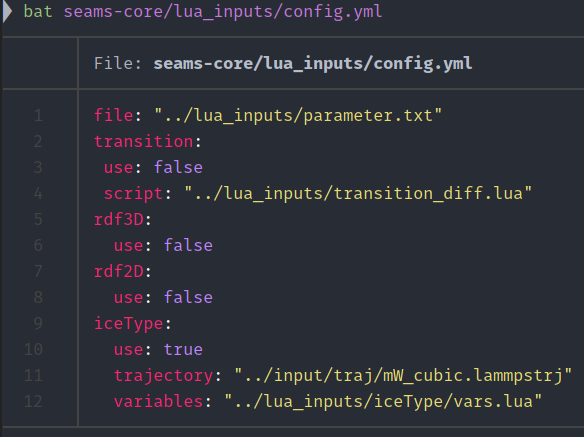
Image Overview
Some sample images encountered during use.
Get The Code
Contributors welcome! As a FOSS project, we welcome contributions in any form, especially documentation and analysis methods.
Get in touch
We prefer GitHub issues. However, for joint research endeavors, use the form below.
© d-SEAMS core team. All rights reserved. Design: HTML5 UP. Adapted by Rohit Goswami (HaoZeke)